At the May, 2005 annual meetings of the American Society of Clinical Oncology (ASCO), Cree and associates presented their long-awaited findings from a study that began in the late 1990s. 81 patients received chemotherapy selected by the results of a cell death assay (ATP endpoint) and 78 patients received "clincian's choice" chemotherapy. There was a 42% response rate in the assay-directed group, compared to a 34% response rate in the clinician's choice group (not statistically significant or "n.s." - click here for tabular view of response data). There was a 68% response + stable disease rate for assay directed therapy, compared to 54% for clinician's choice (n.s.). There was a slight trend for improved progression free survival (which was 104 days; hazard ratio 0.8, confidence interval 0.6 - 1.1; median ratio 1.12). There was no difference in overall survival (HR 1.01), with a median survival in both arms of just under 9 months. A link to the lead investigator's complete presentation (requires Windows Media Viewer) is provided elsewhere on this website.
Weisenthal's
comments:
Firstly, Cree, Kurbacher,
and associates are to be commended for doing what has never been done
before
in the long history of cell culture drug resistance testing (CCDRT),
which
is to successfully complete a prospective, randomized trial to
determine
if treatment outcomes may be improved through the use of CCDRT. A
number of investigators (including me) have tried to organize and
complete
such trials, only to have the trials closed prematurely because of poor
patient accrual and/or other factors, such as low assay evaluability
rate
(no longer a problem with the newer, cell death endpoint assays).
The successful completion of a prospective, randomized trial is a
landmark
achievement.
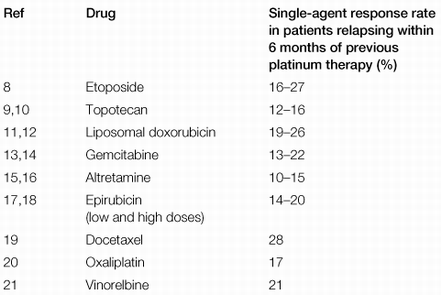
The results were impressive with respect to response rates, but disappointing with respect to overall survival. There was a 20% improvement in response rates with assay-directed therapy, which likely would have achieved significance, had the study been adequately "powered" (i.e. had sufficient numbers of patients been enrolled to allow for the observed differences to have become statistically significant). What was quite impressive were the response rates in both arms of the study, which exceeded those previously reported in other studies which enrolled at least 80 patients per treatment arm (click here for a summary table of response rates in platinum-resistant ovarian cancer previously reported in the literature; typically about 20%, compared to the 34 - 42% response rates in the Cree study).
However, the improvement in progression-free survival (the primary study endpoint) was so marginal that it would have likely required several hundred patients in each arm of the study to achieve significance, presuming that there was a real difference, and the absolute magnitude of benefit (10% improvement in median relapse-free survival ratio) was modest, indeed. Finally, no advantage in overall survival was seen, although the study did allow the clinician's choice patients to be "crossed over" to assay-directed therapy upon disease progression. (Many of these latter patients did receive assay-directed therapy, with a quite astonishing 40% response rate to what was then third-line therapy). Still, median overall survival for all patients was less than 9 months, and the percentage of patients surviving three years was only about 10%.
I believe that there are a number of important caveats and lessons, however (some pointed out by Cree, et al themselves; some of which are new to the present discussion).
I'd like to begin by making some remarks about platinum-resistant ovarian cancer in general and then to present a brief description and analysis of our own experience with this disease.
"Platinum-resistant" disease is most commonly defined as either disease progression during platinum-based chemotherapy or else disease recurrence occuring within 6 months of the discontinuation of platinum-based chemotherapy.
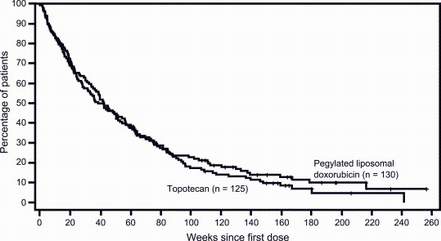
The next linked figure shows a typical clinical trial result (overall survival) in"platinum-resistant" ovarian cancer: minimal difference between the two arms of the study (Doxil versus topotecan) and an 8-12 month median survival. In contrast, with so-called "platinum-sensitive" ovarian cancer, significant differences in overall survival between treatment arms are sometimes observed (e.g. as with Doxil versus topotecan, click here) and median survivals are twice as long as compared to "platinum-resistant" ovarian cancer.
So problem number one for Cree, et al, was the unfavorable biology of platinum-resistant ovarian cancer. It's a disease where no form of chemotherapy has ever been shown superior to any other form and where chemotherapy hasn't been shown to produce survival advantages compared to no chemotherapy. They'd certainly have had a better chance of "success" by performing their trial in either platinum-sensitive ovarian cancer or previously-untreated ovarian cancer, where it is more apparent that chemotherapy improves the survival of populations of patients and where identifying the most effective chemotherapy would be more likely to improve survival above that achieved with empiric chemotherapy.
Note also that even relatively small studies, such as the above, have typically enrolled many more patients than were entered onto the Cree study (which required about 4 years to accrue 160 patients). It's likely that statistical significance for at least response rate would have been achieved by Cree and associates, had they enrolled twice as many patients; however, this would have taken another 4 years, during which treatment advances, such as the introduction of new drugs. could well have rendered the Cree trial obsolete and irrelevant. So problem number two was the small size ("underpowering") of the Cree study.
Another interesting point, discussed by Dr. Cree in his ASCO presentation, is the presence of a "learning" effect, in which clinicians saw the success of the combinations selected by the ATP assay and tended to use these combinations in the later years of the study (when most patients were accrued), instead of using more conventional drug regimens in use at the time of the study. In the first year of the study, the study oncologists tended to use traditional 2nd line treatment regimens. Progression-free survival for patients entered during the first year was significantly better in the assay-directed treatment arm than in the clinician's choice treatment arm. Thereafter, the study oncologists began to choose the novel drug combinations which were being selected in the assay-directed arm, and treatment outcome differences between the two arms narrowed.
Also, there was little heterogeneity in the therapies chosen by the assay, with 2/3 of the patients being identified to receive just two regimens (gemcitabine/treosulfan or mitoxantrone/paclitaxel) and virtually all patients being identified to receive just three regimens (the preceding two, plus cisplatin/gemcitabine). Overall, there was little difference between the distribution of regimens given in the assay-directed arm of the study, compared to the clinician's choice arm of the study. Thus, problem number three is that there wasn't really all that much difference in the treatments received in both arms of the study (particularly when, upon disease progression, patients in each arm of the study could crossed over to the treatments of the other arm of the study). Although the cell death (ATP endpoint) assay clearly gets the "credit" for discovering two of the novel drug combinations which produced the high response rates, it wasn't established that matching patient to drug combination on an immediate (rather than deferred) individual basis improved the treatment outcomes.The next linked table shows the distribution of the drug regimens given in both clinician's choice and assay-directed arms of the study.
Problems number four and five relate to the fact that the patients on the Cree study constituted an unusually poor prognosis group of patients. This is underscored by the results of two other studies presented at the same (May, 2005) ASCO meeting (Harter, et al. , Abstract # 5004 and Crawford, et al., Abstract # 5003).
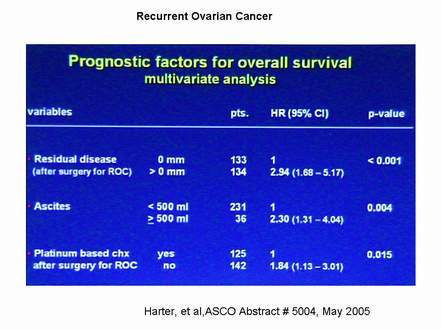
Harter, et al (Abstract # 5004) examined prognostic factors in recurrent ovarian factor. They found that the only significant factors on multivariate analysis were (1) incomplete surgical resection of all recurrent disease, P<0.001, click here; (2) presence of substantial ascites (P=0.004, click here), and (3) lack of platinum-based chemotherapy for recurrent disease (P=0.015; note that the Harter study included both platinum-resistant and platinum-sensitive disease and that the choice of platinum for second line therapy was probably a marker for patients with platinum-sensitive disease).
Crawford, et al (Abstract #5003) compared ovarian cancer surgical practices in the United Kingdom (from where most of the Cree/ATP assay directed therapy study were recruited) with surgical practices in other countries. Crawford reported that the UK patients were far less likely to have received complete surgical resection than those patients treated in other countries (e.g. see linked graph). The Crawford study had to do with previously untreated patients, and it is very likely that UK patients with recurrent disease receive even lesser degrees of surgical resection.
With regard to the reports of Harper et al and Crawford et al, it is notable that only about half of the Cree, et al patients had received a prior hysterectomy/oopherectomy, which is standard surgical treatment in most of the world. Cree, et al, did not state the percentage of patients who had received a total resection of recurrent disease, but it is doubtful that many of their patients received this. Finally, about 2/3 of their specimens tested were from ascites fluid (understandable in a country where recurrent ovarian cancer is not generally treated with surgery and where the ability to obtain tumor cells for testing would therefore be skewed toward patients with large volume ascites). Thus, the Cree study population was unfavorably skewed with regard to the two most important prognostic factors in recurrent ovarian cancer.
At this point, I would like to compare our data for platinum-resistant ovarian cancer with that of Cree, et al. Our data, as will be seen, are considerably more encouraging than were those of Cree, et al. I think that there are two likely reasons for this difference: firstly, differences in patient prognostic factors and secondly, differences in assay methodology.
The following linked figure shows the overall survivals of ovarian cancer patients who had tumor specimens submitted to our laboratory for testing within 6 months of non-neoadjuvant, platinum-based chemotherapy. 55 patients with solid tumor biopsies and resections had a median survival of 19.5 months, compared to 5 months for 4 patients with solid tumor resections but inevaluable assays, and 6.6 months for 10 patients for whom we tested ascites fluid. Differences between solid tumor biopsies and fluids were not significant (P=0.10), but the difference between solid biopsies successfully tested on one hand and the aggregate total of fluid specimens and unsuccessful solid tumors on the other hand was significant (P<0.05). The median survival of all 65 patients with evaluable assays was 18.0 months and the median survival of all 69 patients ("intention to treat") was 17.1 months. These survivals compare favorably with both the existing clinical literature and with the Cree study.
Two of the most likely reasons for the difference between patients with tumors submitted to our laboratory for testing and the Cree patients were (1) markedly lower representation of patients with ascites among our patients and (2) much more aggressive approach to surgery (the most important treatment modality in ovarian cancer) in the US compared to the UK. However, there are other differences, as well.
The next linked table once again shows the drugs tested and regimens given in the Cree study. In the Cree study, only 12 drugs and combinations were tested. One of the reasons for this is that the Cree methodology uses a wide range of drug concentrations to determine an in vitro drug resistance score, calculated from results at all the concentrations. Because of the need to test many drug concentrations, comparatively few drugs can be tested per specimen. In contrast, our approach has been to identify a single, most important "index" drug concentration (the concentration which produces the greatest standard deviation or "scatter" with respect to drug-induced cell death between specimens) and to test the cells only against two drug concentrations, the "index" concentration and 1/2 the "index" concentration. Our biological and clinical correlations are better when we test the most informative drug concentration than when we test a broad concentration range and interpolate or extrapolate a result. This approach is also advantageous, in that it allows us to use the (sometimes limited) quantity of tumor cells to test a larger number of drugs and to test the drugs with at least two different cell death endpoints (which are mutually complementary).
The next linked table shows the in vitro best regimens for our 75 patients with platinum resistant ovarian cancer (some of these patients did not have sufficient follow-up to be included in the above survival data or follow-up information was not available). Three factors are particularly evident, in comparison to the Cree study. Firstly, there is a much broader range of agents and combinations tested and, secondly, there was a much broader range of in vitro best regimens selected by the assay results. Thirdly, in about 10% of cases, all of the drugs and regimens tested were in the "resistant" range and no clear cut in vitro best regimen could be identified. In the past, there has been the feeling that all patients must or should be offered treatment with cytotoxic chemotherapy. However, the situation is NOT that either chemotherapy works or it doesn't. The true situation is that ineffective chemotherapy can diminish not just quality of life but also quantity of life, through organ toxicity, immunosuppression, and by inducing mutations in genetically unstable tumor cells to a more aggressive phenotype. The truth of this last statement is increasingly supported by studies in diseases such as metastatic breast cancer, where the use of more complex and aggressive therapy increases response rates and increases survival in some patients, while not increasing overall survival, meaning that empiric chemotherapy is often a "zero sum game," wherein some patients have their lives extended, while others have their lives shortened. Thus, identifying patients with refractory neoplasms may not only spare them toxicity but may prolong their lives, by sparing them from the life shortening effects of ineffective chemotherapy. Such patients may be more likely to benefit from less toxic forms of therapy, such as angiogenesis inhibitors, high dose tamoxifen, EGFR antagonists, clinical trials, or supportive care.
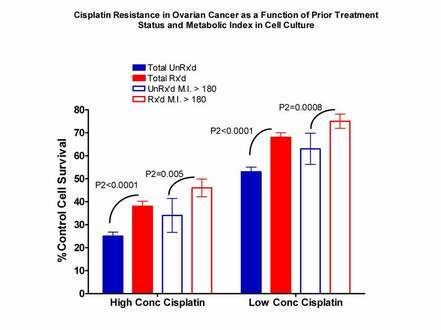
As a final point relating to assay methodology (and this point is highly technical and likely to be appreciated only by workers in the cell culture drug resistance testing field, to whom this is directed), I believe that it is very important to adjust for tumor "robustness" and three dimensionality (in culture) when calculating and interpreting assay results. For example, the attached chart (click here) shows a comparison between assay results with cisplatin, tested against (1) specimens from previously-untreated ovarian cancer patients and (2) previously-treated patients. As noted, we test drugs at only two concentrations, a "high" concentration (in the case of cisplatin, 3.3 ug/ml) and a "low" concentration (1.65 ug/ml). It can be seen that specimens from previously-treated patients have significantly greater resistance to cisplatin than specimens from previously-untreated patients. However, when we examine only a subset of specimens (those with a high "metabolic index," defined as the Day 4 (end of culture) viable tumor cell number divided by the Day Zero (pre-culture) viable tumor cell number, multiplied by the Day 4 control MTT formazan signal (which indexes mitochondrial Krebs cycle activity)), the specimens from previously untreated patients with a high metabolic index appear to be just as resistant to cisplatin as are specimens from previously-treated patients from the entire dataset. However, when the high metabolic index specimens from previously-untreated patients are compared with the high metabolic index specimens from previously-treated patients, the latter specimens are significantly more resistant than the former, just as expected. Thus, it is important to compare "apples to apples" in classifying assay results with respect to "sensitivity" versus "resistance" to the different drugs. This is a very complex topic, which I'd be happy to discuss in greater detail with individual investigators in this field, upon request.
I want to close by making a comment on the planned Gynecologic Oncology Group study in platinum-resistant ovarian cancer, utilizing the Oncotech "EDR" assay to direct chemotherapy.
I am intimately familiar with the "EDR" assay, as I was the one who identified it and even named it, when I was one of the two co-founders and original Laboratory Director and chief science officer at Oncotech (it's a long story, which I'll save for another time). This assay is specifically designed to identify INACTIVE drugs and the Oncotech reports even state that the assay results should NOT be used to identify ACTIVE drugs. A pilot study, in which two GOG investigators used the Oncotech EDR assay to identify the "best" treatment regimens for platinum-resistant ovarian cancer was negative, with both assay directed and non-assay directed patients having the same median survival, which were 13 and 12 months, respectively (for reference and discussion, click here).
I have repeatedly tried to get the Gynecologic Oncology Group to allow me to donate our services free of charge for a clinical trial of assay-directed therapy in platinum-resistant ovarian cancer, to no avail. The Society for Gynecologic Oncology refused to allow us to present our data at their annual meeting in 2003 and even refused to publish our submitted abstract in their meeting Proceedings (click here to view abstract). One GOG study chairman stated that they will only consider proposals from GOG "members." Another GOG study chairman wouldn't even answer my correspondence (click here to read the letter I sent to him).
I have been trying to do studies with the GOG since 1992, when I and Dr. Robert Nagourney first proposed a head to head to head comparison of the Oncotech EDR assay with the DISC (cell death assay) and the ATP (another cell death) assay. They declined to do the study, in favor of performing an Oncotech-funded study which was never published.
The same investigators who maintain that assay directed therapy should not be used until "proven" in prospective randomized trials are the same people who control the clinical trials system, the grant review study sections, and the journal editorial boards, who have, through the years, refused, denied, and rejected any and all efforts I've made to get these trials off the ground.
Catch 22.
Larry Weisenthal
Revised August 11, 2005
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}