Comments from NSCLC patient received February, 2004
Question relating to the use of the DISC assay in testing Biologic Response Modifiers received 9/2/2002
Letters received pertaining to ONCOLOGY paper "Current Status of Cell Culture Drug Resistance Testing (CCDRT) Click here for full text of paper
Question from European PhD student, regarding the suitability of the MTT assay for testing taxanes (docetaxel and paclitaxel) received 8/6/2002
Additional miscellaneous letters
Subject: Timing of biopsy relative to timing of ongoing cancer chemotherapy and/or radiation therapy
Question:
Date: 1/31/02 5:21:19 PM Pacific Standard Time
From: JonNNNN@aol.com
To: mail@weisenthal.org
I found your site listed on www under chemosensitivity. Currently, I am undergoing radiation and chemotherapy for breast cancer.
Some labs indicate that it is OK to send biopsy immediately for tests without affecting test results. Other companies say I must wait 3 weeks after any treatment. How do I find out which of these opinions is correct? My literature search proved futile. Is there anything in print? Thanks.
Jon NNNNN
Answer:
Date: 1/31/02 5:55:37 PM Pacific Standard Time
From: mail@weisenthal.org (Larry Weisenthal MD)
There are no published data; nor do I believe that there are any useful unpublished data. There is only opinion. I'm not sure that you asked, but here's mine:
I'd prefer to have a minimum of 3 weeks elapse between chemotherapy and/or radiotherapy (assuming that the radiotherapy is being delivered to the area where the tumor will be biopsied...if the tumor to be biopsied lies outside the radiation therapy port, then radiation makes no difference to the timing of the biopsy). The theoretical problem is that, when one tests tissue which has been removed shortly after (or during) chemotherapy/radiation therapy, then all of the drugs one is testing in the assay are really being tested as a combination...a combination of the drug being tested and the chemotherapy/radiotherapy already given to the patient.
The second disadvantage (particularly for a proliferation-based assay; less so for a cell death based assay) is that the tumor cells are probably less likely to proliferate in the culture if they have been removed shortly after chemotherapy/radiotherapy; thus, one is less likely to obtain a high quality assay or even to obtain an evaluable assay.
That said, sometimes there is no choice...as in the case where chemotherapy/radiotherapy is given and then there is the need for surgery (as in the case of neoadjuvant therapy or in case of some surgical emergency). I n these cases, the tumor is being removed anyway and there may be no other source of tumor to biopsy 3 weeks down the road; so it may be worthwhile to proceed with the testing, despite the above theoretical disadvantages.
If you can tell me more about the specific situation in question, I can maybe provide a more specific answer.
Best wishes,
Larry Weisenthal
__________________________
Is one "sensitive" drug better than another? Can you detect drug synergy? What are the best drug combinations?
Question:
In a message dated 2/4/2002, NNNN writes
<< Suppose your assays come up with a bunch of drugs that are all good at killing tumor cells, and let us say that that the patient is someone like the leukemia patient we spoke about, where past treatment has shown to be very good at killing tumor cells in vivo, only to have the tumor cells return very quickly after treatment stops. If a patient's tumor cells are killed by a variety of drugs, does that mean that all drugs worked equally well to attain those results (I would think not)? Is there some some way that synergy can be predicted between drugs that appear to work well individually? Are very complex drug combinations ever advantageous?
Answer:
Date 2/4/02
From mail@weisenthal.org (L Weisenthal MD)
Chemotherapy fails the patient by different mechanisms. Most often, just because of intrinsic resistance of the tumor cells to the drug(s) in question. Sometimes, because of what is called "schedule resistance." The drugs nicely knock down the number of tumor cells, but the tumor grows so fast that it comes back, bigger than ever, by the time the patient has recovered from the effects of the first chemotherapy and is now ready to receive the next dose. This latter mechanism (schedule resistance) is pretty common in diseases such as acute leukemia and certain types of non-Hodgkin's lymphoma, unfortunately.
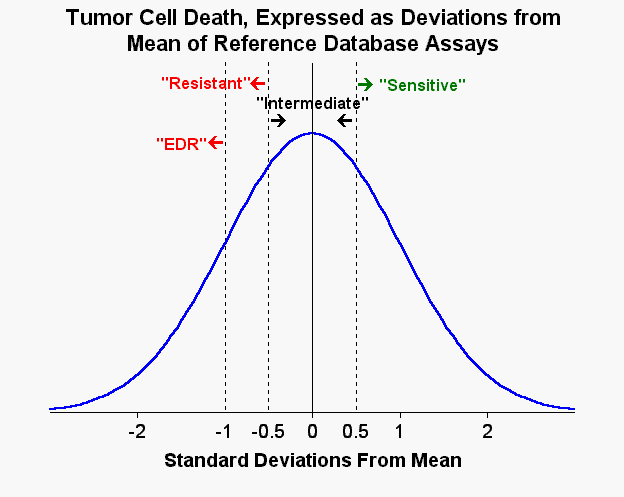
With regard to the other questions, I try not to make the assays do more than that which they are capable of doing. The assays are pretty darn good at distinguishing between ineffective drugs and at least moderately effective drugs. They aren't as good at taking a group of "effective" drugs and picking out the one or two drugs which are most effective. In practice, I always do try to identify what I consider to the the "in vitro best drug." Let's say that one "sensitive" drug is barely to the right of the "Sensitive"/"Intermediate" cut - off ...
e.g. see http//www.weisenthal.org/gaussian.gif (then click "return" arrow on browser to come back)
...while another drug is way over to the right (in other words 67th percentile vs 99th percentile). Logically and intuitively, the 99th percentile drug would be better than the 67th percentile drug...but that's all there is, logic and intuition, as neither I nor anyone else really has the clinical correlation data to define something analogous to "extreme drug resistance (EDR)" -- in this case "extreme drug sensitivity(EDS)" -- and to prove that "EDS" is acually better than just "S."
With regard to the question of synergy; yes, this can be and is observed in both the clinic and in our assays. Unfortunately, true synergy is rather uncommon in most adult solid tumors. Most drug combinations in diseases such as lung cancer, breast cancer, and ovarian cancer are merely additive (whole equals the sum of its parts) and not synergistic. In hematologic neoplasms (leukemia, lymphoma, multiple myeloma), true synergy is very common, both in the clinic and in our assays. In cases where drugs are only additive and not synergistic, we don't learn anything additional by testing the drugs in combination over what we learn by testing them separately. So I only test drugs in combination in cases where I know that there is the realistic possiblity of seeing true synergy.
Such solid tumor drug combinations as cyclophosphamide plus doxorubicin or carboplatin plus Taxol are virtually always only additive and not synergistic. Cisplatin plus etoposide is virtually never synergistic in non-small cell lung cancer, but is synergistic 25% - 50% of the time in small cell lung cancer. Gemcitabine plus cisplatin is very often synergistic. Vinorelbine plus mitomycin c or mitoxantrone is/are occasionally synergistic, as is irinotecan plus cisplatin. And so on.
With respect to the question of highly complex (3, 4, or 5 drug) combinations, the problem is that one must often reduce the dose of the individual drugs, to avoid intolerable toxicity. So the question is whether or not it is better to give multiple drugs at lower doses or fewer drugs at higher doses. The best combinations are those in which there is true synergy and in which the toxicities of the drugs in the combination are non-overlapping, so that full doses of each drug may be given safely. There are relatively few situations where this ideal is met, unfortunately.
__________________________
Request for "nuts and bolts" technical information on how to perform Human Tumor Assays:
Question:
December 6, 1999
Dear Dr Weisenthal, I have visited your websites several times in the hope of understanding the techniques of in vitro drug sensitivity testing for cancer, the information was very useful. We are currently planning some research in to cancer chemotherapy using such techniques. I write to ask if you know of any workshops or courses that are being conducted anywhere in the world for scientists who wish to set up such facilities. I would appreciate any information that you can give me
With regards (Name and institution withheld) Senior Scientist Research and Administrative Unit
Answer:
Dear Dr. ______ You make a very good suggestion. 20 years ago, Dr. Sydney Salmon of the U of Arizona held two international workshops to teach people how to perform the "Human Tumor Stem Cell Assay." This was a clonogenic, or colony-counting, assay which did not prove to be useful for most applications. More recently, Drs G.J.L. Kaspers and A.J.P. Veerman have held two workshops at the Free University of Amsterdam on the use of different assays in hematologic neoplasms. The first of these was published as a book: Kaspers GJL, Pieters R, Twentyman PR, Weisenthal LM, and Veerman AJP (eds). Drug resistance in leukemia and lymphoma. Harwood Academic Publishers Langhorne PA (USA), Tokyo (Japan), Amsterdam (Netherlands), Paris (France), Berlin (Germany), Reading, Berkshire (Great Britain), and Victoria (Australia) copyright 1993
With the belated recognition of the importance of the cell death endpoint (measured by the DISC, MTT, ATP, and Fluorescein diacetate assays ... all of which provide substantially similar results)....it probably would be time to hold a such a workshop in solid tumors. My own plans are to devote the next year to writing up papers on the data I've been accumulating for the past 8 years. Perhaps in the year 2001 or 2002 I shall try to organize a workshop. In the meantime, you might also correspond with Dr. Andrew Bosanquet in Bath, England and Dr. Robert Nagourney in Long Beach (Rational Therapeutics). Links to their web sites appear at the bottom of "The Human Tumor Assay Journal" website.
The following website also gives useful technical information on specimen preparation and culture. Although the site is maintained by the manufacturer of an instrument to read the results of the fluorescein diacetate assay (FDA, FMCA), this information provides step by step details which may be applied to any cell death assay system (FDA/FMCA, DISC, MTT, and/or ATP).
I do have 2 important suggestions. First, it is important to culture solid tumor specimens in conical polypropylene microwell plates. Polypropylene is a slippery material which, in most cases, prevents the attachment of fibroblasts and epithelial cells and encourages the tumor cells to remain in the form of 3 dimensional, floating clusters. Secondly, the linked website below provides a protocol for culturing cells for 72 hours. This is because the assay described (the FMCA) was optimized firstly for hematologic neoplasms and then just adapted for solid tumors. But I think it is much better to culture solid tumors for 96 hours before assessing drug effects, because there is often substantially greater loss in the viability of normal connective tissue and normal epithelial cells, relative to tumor cells, at 96 hours, compared to 72 hours. Endpoints such as MTT, ATP, and fluorescein diacetate cannot discriminate between tumor cells and normal cells (this distinction can be made, however, with the DISC assay). Therefore, it is important that the preponderance of viable cells at the end of the culture be tumor cells and not normal cells. Culturing cells in conical polypropylene microwells and culturing them for 96 hours increases the proportion of tumor cells, relative to normal cells.
On the other hand, a 72 hour culture period does make a lot of sense for assays on lymphatic neoplasms (NHL, CLL, ALL, and myeloma), where there is often substantial attrition of neoplastic cells between 72 hours and 96 hours.
http://www.labsystems.fi/instru/manuals/applicat/an201.htm
Best wishes, Larry Weisenthal
__________________________
{kind=link}